2014 is almost over, my friends. The year is coming to a close, and like every year—but probably plenty more this year, thanks to Buzzfeed—a barrage of “Best of” lists will soon besiege us. Already, we have had the best subreddits, the best movie posters, and the best wine books of 2014 enumerated for us. But what I am really looking forward to this year is the Best Newly Discovered Diseases of 2014. Of course, we will probably see mucociliary clearance disorder (from CCNO mutations) and chronic atrial intestinal dysrhythmia syndrome (from a SGOL1 mutation), but will PGM1 deficiency make the list? Phosphoglucomutase 1 (PGM1) is the gene of the week, but will we remember it as one of the important genes of this year?

To be clear, PGM1 deficiency has been around for a while—since the 1960s!1—but only as a glycogenosis, or glycogen storage disease (GSD), a defect in the synthesis and breakdown of glycogen. Glycogen is a polysaccharide made up of a central glycogenin protein, surrounded by glucose, which is released to provide energy throughout the body. PGM1 deficiency, the GSD, was known for its exercise intolerance (a.k.a. my excuse for not going to the gym more often), which could be partially treated with glucose infusion2.

Glycogen looks crazy! This is glycogenin surrounded by several thousand glucose units! (Image from Wikipedia)
In February, however, Tegtmeyer et al.3 further characterized PGM1 deficiency as a “congenital disorder of glycosylation,” observed in 19 patients from 16 different families, thus making it a novel disease, as far as I’m concerned. Glycosylation is the process in which a carbohydrate (“glycan”) is attached to another molecule, such as a protein or a lipid. Protein N-glycosylation occurs when glycans are attached to asparagine residues of proteins. N-glycosylation is an important cotranslational or posttranslational step necessary for the proper folding and stability of some proteins. Glycoproteins (the sugar-protein hybrids that result from N-glycosylation) are often secreted or reside in the cell membrane, where they are important for cell-cell adhesion. Congenital disorders of glycosylation (CDGs) are defects in protein N-glycosylation.
CDGs can come in two varieties: Type I, in which synthesis of the glycan precursor is impaired, and Type II, in which modification of the protein-bound glycan is impaired. Interestingly, Tegtmeyer et al. found that PGM1 deficiency patients exhibited elements of both CDG-I and CDG-II. See the figure below for details!
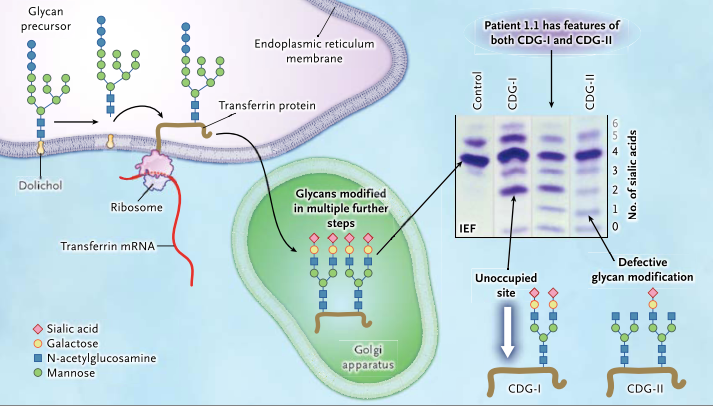
A glycan precursor is assembled from monosaccharide units on dolichol in the membrane of the endoplasmic reticulum. Each glycan precursor is then transferred to a specific asparagine residue in the nascent peptide chain of a protein being synthesized by a ribosome and entering the endoplasmic reticulum. Because transferrin (brown line) is abundant in blood, it is used as an indicator of the efficiency of the glycosylation system. The protein is transported to the Golgi apparatus, and the glycans are modified in multiple steps. If a genetic defect impairs synthesis of the precursor glycan in the endoplasmic reticulum, one or both glycosylation sites of transferrin may remain unoccupied (white arrow). Such defects are classified as congenital disorders of glycosylation type I (CDG-I). In congenital disorders of glycosylation type II (CDG-II), the defect occurs in the modification of the already-transferred glycan. Isoelectric focusing (IEF), which separates molecules according to differences in their isoelectric point, is used for investigating congenital disorders of glycosylation with the use of an agarose-gel–based system, followed by immunoprecipitation in gel with an antibody to transferrin. Under conditions of normal glycosylation, most transferrin molecules carry four sialic acid residues and form a single major band on IEF. A few transferrin molecules carry five or six sialic acid residues that appear as minor bands on IEF. CDG-I defects are easily identified owing to the occurrence of transferrin isoforms with two or no sialic acid residues detected by means of IEF. No signal for monosialotransferrin (transferrin with one sialic acid residue) is seen in such disorders. In CDG-II, the defect can result in transferrin molecules with no, one, two, three, or four sialic acid residues. A patient with a CDG-II defect typically has bands of approximately equal intensity at all positions from 0 through 3 or a smooth gradient of intensities, whereas in most cases, tetrasialotransferrin shows a more intense signal owing to residual activity. The patients in this study had features of both CDG-I and CDG-II, as shown by IEF, suggesting a novel disorder. (Figure and modified caption from Tegtmeyer et al., 2014)
Although all PGM1 deficient patients had bifid (split or cleft) uvula at birth, additional phenotypes varied greatly. Phenotypes of GSDs and CDGs include hepatopathy, hypogonadotropic hypogonadism, growth retardation, hypoglycemia, myopathy, and cardiac arrest. The mechanisms leading to the development of these clinical manifestations are unclear, but Lee et al.4 recently published the effects of specific PGM1 mutations on PGM1 protein function. Some mutations cause impaired PGM1 folding and solubility while others impair its enzymatic activity (or both). Lee et al. found that mild biochemical impairments correlated with milder clinical phenotypes.
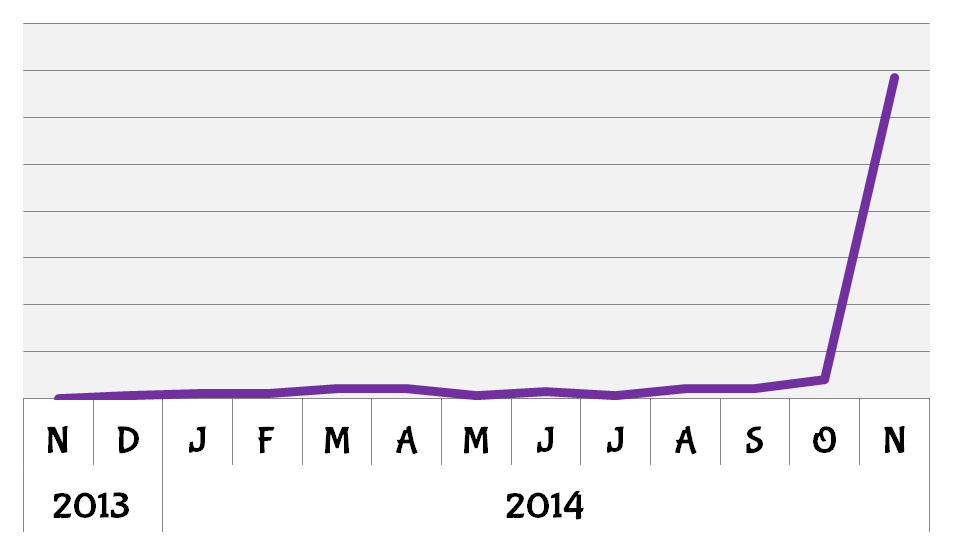
2014 was a good year for PGM1. BioGPS queries for the gene increased sharply following the publication by Lee et al. on the mechanisms by which mutations in PGM1 can effect its function and lead to the clinical features of PGM1 deficiency.
It will be interesting to see how the findings from Lee et al. will lead to better prognoses and treatments for PGM1 deficiency. In the meantime, Tegtmeyer et al.5 have already tested out a treatment for the disease. To me, the coolest part about the discovery of PGM1 deficiency as a CDG was the discovery by Tegtmeyer et al. (in the same publication!) that dietary supplementation with galactose could improve glycosylation and related clinical features in patients. Within a few weeks of treatment, rhabdomyolysis subsided and hypogonadotropic hypogonadism resolved in two of six patients.
So, even though PGM1 deficiency is not a new disease by name, PGM1 deficiency as a CDG is new. Futhermore, the expedient publication by Lee et al. on the mechanisms behind the new PGM1 deficiency, as well as potential treatments suggested by Tegtmeyer et al., make 2014 a great year for PGM1 deficiency: my nominee for best new disease of 2014.

References:
- Tegtmeyer, L. C., Rust, S., van Scherpenzeel, M., Ng, B. G., Losfeld, M.-E., Timal, S., Raymond, K., et al. (2014). Multiple phenotypes in phosphoglucomutase 1 deficiency. The New England journal of medicine, 370(6), 533–42. doi:10.1056/NEJMoa1206605 [↩]
- Preisler, N., Laforêt, P., Echaniz-Laguna, A., Ørngreen, M. C., Lonsdorfer-Wolf, E., Doutreleau, S., Geny, B., et al. (2013). Fat and carbohydrate metabolism during exercise in phosphoglucomutase type 1 deficiency. The Journal of clinical endocrinology and metabolism, 98(7), E1235–40. doi:10.1210/jc.2013-1651 [↩]
- Tegtmeyer, L. C., Rust, S., van Scherpenzeel, M., Ng, B. G., Losfeld, M.-E., Timal, S., Raymond, K., et al. (2014). Multiple phenotypes in phosphoglucomutase 1 deficiency. The New England journal of medicine, 370(6), 533–42. doi:10.1056/NEJMoa1206605 [↩]
- Lee, Y., Stiers, K. M., Kain, B. N., & Beamer, L. J. (2014). Compromised catalysis and potential folding defects in in vitro studies of missense mutants associated with hereditary phosphoglucomutase 1 deficiency. The Journal of biological chemistry, 289(46), 32010–9. doi:10.1074/jbc.M114.597914 [↩]
- Tegtmeyer, L. C., Rust, S., van Scherpenzeel, M., Ng, B. G., Losfeld, M.-E., Timal, S., Raymond, K., et al. (2014). Multiple phenotypes in phosphoglucomutase 1 deficiency. The New England journal of medicine, 370(6), 533–42. doi:10.1056/NEJMoa1206605 [↩]