With coronavirus mutations overtaking the news cycle, fear and uncertainty are spreading as efficiently as the virus itself. Viruses are expected to evolve and mutate. Not all mutations are meaningful. Scientists have already recorded hundreds of mutations across thousands of coronavirus cases. Most mutations do not have a significant effect on infection severity or transmissibility. Scientists are tracking a few mutations, however, that have concerning attributes. These mutations have shown to spread quicker and may have implications for vaccine development.
The terminology surrounding SARS-CoV-2 mutations is being used very loosely, but distinction is important. A strain of a virus has distinct properties and causes a specific immune response. When a virus has developed a set of mutations that causes it to act differently than that of the original strain, it is classified as a variant. Similarly, lineages are often minor accumulations of mutations, forming a different genetic line of the strain.
If researchers identify a SARS-CoV-2 variant that have concerning pathogenic properties, the variant is classified as a Variant Under Investigation (VUI) with a year, month, and number. Experts investigate the level of risk the variant poses and may elevate it to the classification Variant of Concern (VOC). The B.1.1.7 variant, for example, was originally classified as VUI-202012/01, but has since been renamed to VOC-202012/01. Researchers also use the term Variant of Interest (VOI) to note a variant that requires further study to identify whether it has clinical significance.
The key differences between these classifications:
- VOIs and VUIs are variants of increasing prevalence with mutations in biologically important regions but are lacking evidence for increased transmissibility, virulence, and/or immune evasion.
- VOCs are variants which have evidence in the literature of increased transmissibility, worse outcomes, and/or decreased vaccine efficacy.
What is a mutation? Why do they arise?
Viruses are DNA or RNA encased in a protein shell. SARS-CoV-2, the virus that causes COVID-19, is made up of RNA. RNA is made up of nucleotides. Nucleotides provide the code for building amino acids, making up the protein shell. Viruses are constantly replicating themselves. A mutation occurs when a nucleotide prints a typo in the code. In SARS-CoV-2, this only needs to occur in one out of 30,000 nucleotides to produce a mutation.
A mutation can be a single-letter typo or longer deletions or insertions in the virus’s genetic code. A deletion mutation (Δ) occurs when part of the code is left out during replication. A single nucleotide may be deleted or an entire section of a chromosome. An insertion mutation is the opposite – one or more extra nucleotides are added into the sequence. Similarly, a substitution mutation occurs when one nucleotide is switched with another.

With variant S:D614G, for example, the mutation occurs on the virus’s spike proteins, or the part of the protein shell that binds to humans’ cells to infect them. This mutation allows the virus to pass quicker between cells in the host’s body. As it copies itself at a faster pace, this increases the viral load in the host. This is not making infected individuals sicker, but it is making them more contagious to others.
SARS-CoV-2 uses a spike protein (S) to infect human cells. D614G is an amino acid mutation in this protein. In its original form (D614), the 614th amino acid in the protein is aspartic acid (“D”). In the mutated form (D614G), the 614th amino acid is glycine (“G”). S:D614G means ‘spike protein with aspartic acid at position 614 mutated to glycine.’
Most RNA viruses don’t have the proofreading enzyme needed to review each string of RNA for mistakes as it replicates. This causes RNA viruses to experience more mutations; errors build up over a shorter time frame, creating different strains. SARS-CoV-2 does have this rare proofreading feature, however. Nucleotides still produce errors; mutations still happen, but at a slower rate than other RNA viruses like influenza.
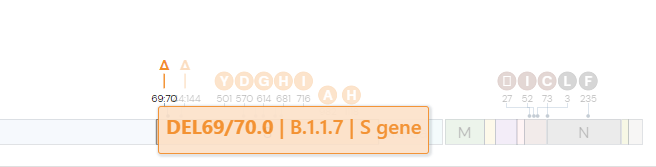
In the deletion mutation S:DEL69/70, the 69th and 70th codons (sequences of three nucleotides) have been deleted. This means the code for the H and V amino acids on the S gene were not printed.
Some of the significant mutations that researchers are currently studying include:
Lineages
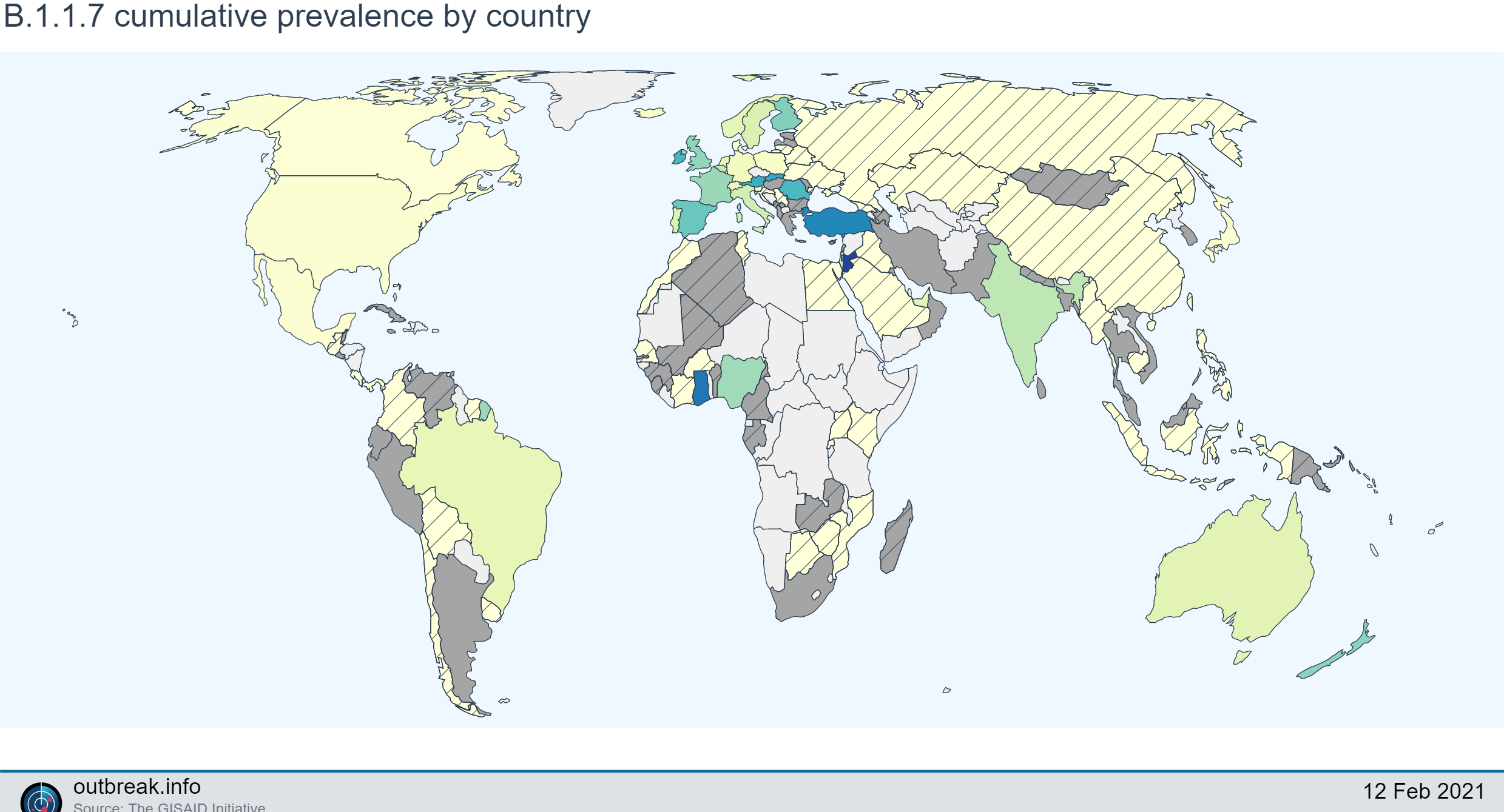
- B.1.1.7 – first identified in the UK in December from a sample that was collected on 9/20/2020 and has since been detected in 55 countries and counting, including numerous US states. This lineage has been shown to have an increased reproduction number, but no evidence for changes in reported symptoms, disease severity, or disease duration.
- B.1.351 – first detected in South Africa on 10/8/2020 and has since in over 30 countries, including the US. Evidence suggests mutations in the lineage may be better at escaping antibodies; however vaccine developers are working on strain-specific booster shots to enhance immunity to B.1.351.
- P.1 – first identified in travelers from Brazil on 12/4/2020 and has since been detected in the US and other countries. Research has indicated mutations in the lineage may be able to more efficiently evade antibodies than the original strain.
- B.1.429 – (also called CAVUI1) identified in California on 7/13/2020 and has since been detected in other countries.
Mutations
- N501Y – first detected in Brazil on 4/6/2020 and has since been discovered in over 66 countries and numerous US states. This mutation has already been found in 34 lineages, including B.1.1.7 and B.1.351, and has shown to support increased transmissibility of the virus.
- E484K – prevalent in many regions in Brazil. This mutation has been discovered in 59 lineages so far, including P.1 and B.1.351. This is a substitution mutation in which a negatively charged amino acid (glutamic acid) is swapped with a positively charged amino acid (lysine). Evidence suggests this mutation is able to escape antibodies and re-infect individuals who have recovered from COVID-19.
- L452R – first detected in Europe, but presence in California has coincided with a rise in cases the state. Study suggests it may be resistant to some neutralizing antibodies. L452R has been detected in 38 lineages, including B.1.429.
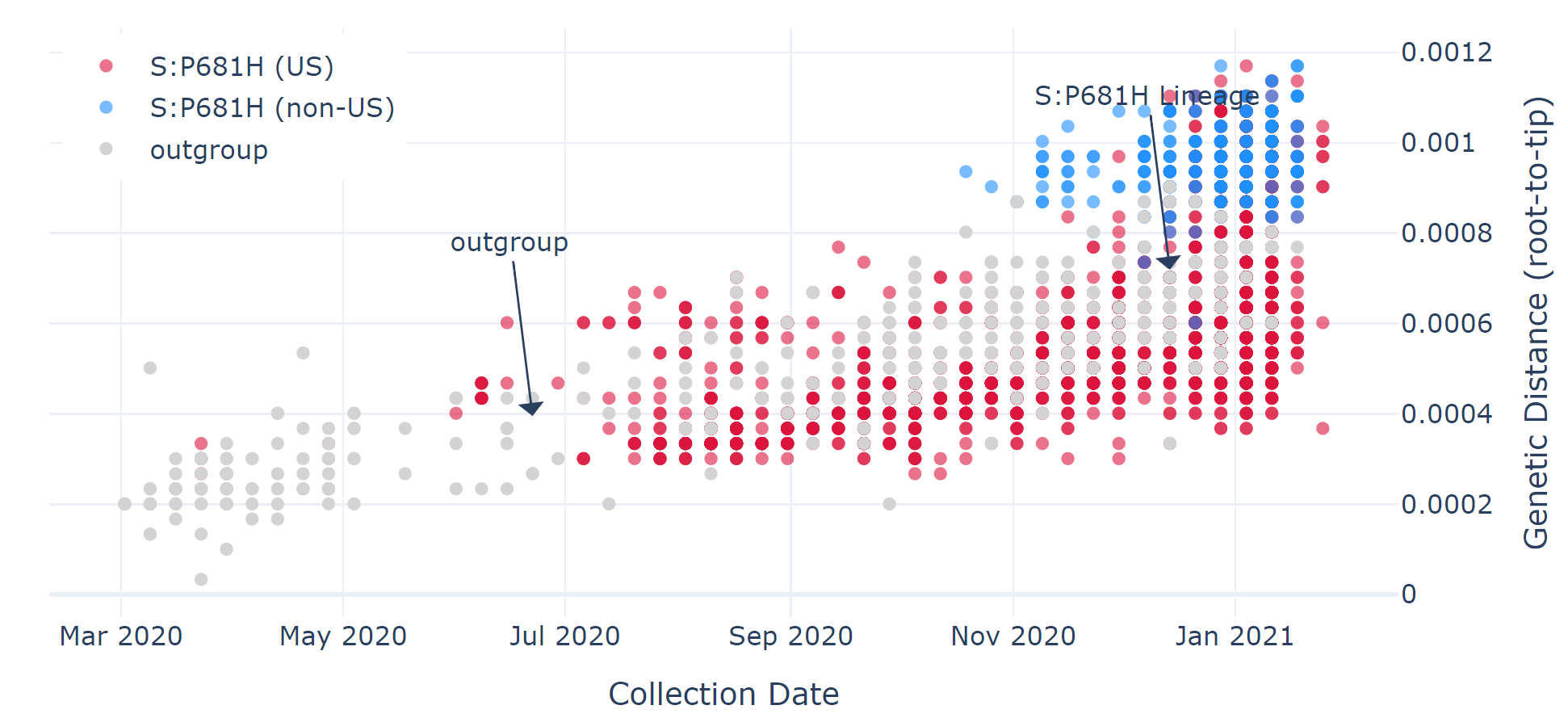
- P681H – first identified in the US, but growing evidence suggests sharply rising global prevalence. This mutation has already been found in 96 lineages, including B.1.1.7.
Researchers around the world have sequenced over 522,000 SARS-CoV-2 genomes and have uploaded the data to the Global Initiative on Sharing All Influenza Data (GISAID) database. The Center for Viral System Biology (CViSB) at Scripps Research has aggregated the information about these mutations in Mutation Situation Reports that are updated daily. Sequencing helps researchers uncover how the virus uses the instructions encoded in its DNA to infect people. Sequencing also helps researchers follow the spread of the virus over time around the globe. This is an important part of understanding how epidemics/pandemics spread and change – and how they can be fought. In a pandemic, information is the most valuable weapon that scientists and the public can wield.
How do these mutations impact the pandemic?
Even when an amino acid in a virus changes and mutation occurs, the virus might not appear any different. Sometimes, mutations can even create viruses that are defective, essentially killing itself right away. Researchers use mutations to trace how the virus is spreading around the world. What is concerning, however, is that some of these variants are now outcompeting the original virus in places like Brazil, the UK, and South Africa.
Researchers around the world are tracking the unusually high mutation rate occurring within a short period of time and observing some repetition in mutation patterns that suggest these variants are not entirely random.
Researchers offer three possible explanations for this:
- an immunocompromised patient experienced a prolonged infection, allowing the virus more time to replicate within a single host, creating more mutations at a faster rate;
- antibodies created selective pressure and the virus is experiencing adaptive mutation or genetic hitchhiking (a gene increases in a population because it sits next to other genes that are beneficial to the virus on the same chromosome);
- or these mutations occurred in animals and were transmitted to humans by zoonotic transmission.
Biomedical researchers are working to uncover more about how the mutations in these strains affect infection, replication, and pathogenesis of the virus. More research is fundamental to understanding how these mutations will impact vaccine development. Moreover, questions remain unanswered about how the transmission rates and reinfection rates of these mutations will shape public health measures. Much like the virus genome, our information changes at a rapid pace. But we have something the virus does not – information sharing, global collaboration, and the tools needed to get ahead. Stay informed with the Mutation Situation Reports from CViSB.
Read more about how CViSB is tracking mutations
Watch researchers discuss data sharing in the midst of a pandemic